Welcome to cisRED
A Comprehensive Resource for Conserved Regulatory Motif Discovery
cisRED is a genome-wide database and analytical platform designed to identify conserved cis-regulatory elements across species. Our mission is to support the global scientific community in understanding the mechanisms of gene regulation by providing high-confidence, computationally predicted regulatory motifs in upstream regions of genes.
With data derived from orthologous genes and co-expressed gene sets, cisRED offers a curated, statistically filtered collection of regulatory sequence motifs likely involved in transcriptional control.
Whether you’re a researcher in genomics, bioinformatics, molecular biology, or systems biology, cisRED empowers you with tools and data to explore regulatory logic at the genomic level.
cisRED (cis-Regulatory Element Database) is an automated system that applies motif discovery algorithms on a genome-wide scale, focusing on conserved regions upstream of transcription start sites (TSS). These regions are known to be enriched with regulatory sequences such as transcription factor binding sites (TFBS). Read more
We currently analyze 1.5 kb upstream of each TSS, excluding repetitive elements, to discover motifs that may control gene expression. The database includes:
- Motif matrices (PFMs/HMMs)

- Gene-motif associations
- Co-expression tables
- Sequence logos and motif groups
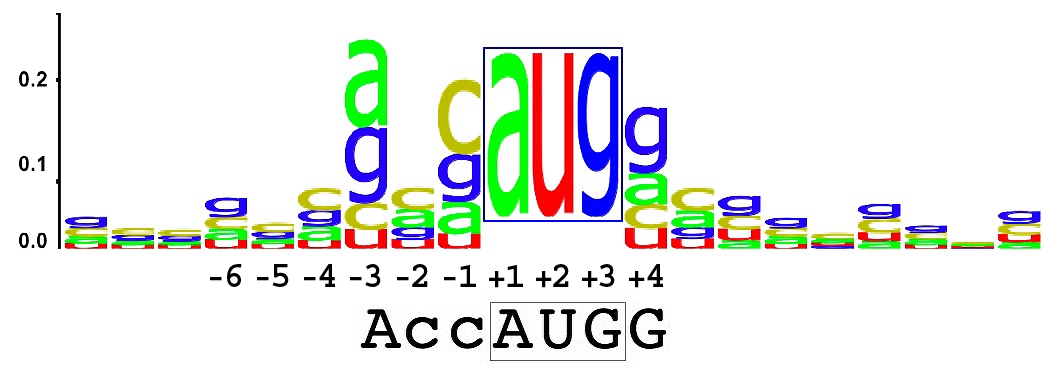
- Integrated genome browser visualization
![]()
To identify biologically relevant motifs:
- Input sequences are extracted from upstream regions of human genes using the Ensembl v22 genome (NCBI build 34).
-
For each gene, cisRED retrieves:
- Co-expressed genes via a custom pipeline
- Orthologues using Ensembl Compara
- Motif discovery algorithms search for statistically over-represented patterns.
- Motifs are filtered using p-values < 0.05, derived from random permutation distributions.
- Results are clustered into motif groups and regulatory patterns, representing possible combinatorial control.
This approach ensures that predicted motifs are evolutionarily conserved, biologically meaningful, and functionally testable.
cisRED is more than just a database. It’s a powerful tool for researchers aiming to understand how genes are regulated at the molecular level. By identifying conserved DNA motifs in promoter regions across multiple species, cisRED enables accurate predictions of transcription factor binding sites essential for genome-wide regulatory analysis.
🔍 Identify transcription factor binding sites
cisRED helps researchers detect conserved DNA motifs upstream of genes, based on co-expression data and orthologous sequences.
🧪 Design ChIP-seq and reporter gene experiments
Motif predictions guide the selection of genomic regions most likely to be involved in gene regulation.
💻 Integrate cisRED data with RNA-seq, ATAC-seq, or custom pipelines
Bioinformaticians use the full dataset to perform genome-wide motif analysis and cross-validate with other omics data.
🌍 Conduct cross-species regulatory element comparisons
Evolutionary biologists study motif conservation to understand how gene regulation evolves across different organisms.
🎓 Support teaching in genomics and systems biology
Educators use cisRED as a real-world example to explain promoter architecture, gene expression control, and regulatory motifs.
cisRED is valuable to:
- Academic and industrial genomics researchers
- Bioinformaticians developing motif discovery pipelines
- Scientists studying gene expression regulation
- Educators teaching functional genomics and regulatory biology
- Students working on promoter analysis and TFBS prediction
- Genome-scale discovery of conserved cis-regulatory elements
- Integrates gene co-expression and evolutionary conservation
- Public access to motif matrices (TRANSFAC, JASPAR) and sequence logos
- Compatible with UCSC Genome Browser and Java-based 3D visualization tools
- Cited in high-impact journals including Nature Methods, Genome Research, and Nucleic Acids
cisRED provides:
🔬 Motif matrices in TRANSFAC, JASPAR, Excel, and plain text formats
📁 Downloadable FASTA input sequences (35 MB)
🗃️ MySQL database schema & dumps (14 MB, public access)
🧬 Sequence logos for every motif in JPG format
🧮 SQL create-table statements for building local instances
🌐 Browser integration with UCSC (Human July 2003 assembly)
🧠 Java-based Sockeye visualization workspace (WebStart)
📈 View upcoming integration with Ensembl genome browser
All data can be used offline by downloading compressed archives from our FTP server.
Title of publication | Summary | Authors & Journal | DOI/Link |
Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing | Introduction of ChIP-seq, combining chromatin immunoprecipitation and high-throughput sequencing to identify STAT1 binding sites in human HeLa S3 cells (stimulated/unstimulated with IFN-γ). Detected ~41,000 STAT1-binding regions. ChIP-seq showed high sensitivity (70–92%) and specificity (≥95%). | Gordon Robertson et al. Nature Methods (in press) | |
cisRED: A database system for genome scale computational discovery of regulatory elements | Describes the development and functionality of cisRED, a database for conserved regulatory motifs. Motifs are predicted in promoter regions using multi-method discovery pipelines across orthologous sequences. Provides atomic motifs, grouped motifs, and motif patterns. | Robertson A.G., Bilenky M., Lin K., et al. Nucleic Acids Research, 2006 | |
Sockeye: a 3D environment for comparative genomics | Presentation of Sockeye, a Java-based application for 3D visualization of comparative genomic annotations. Integrates with Ensembl and supports custom sequence data. Enables visual exploration of genome structure and conservation. | Montgomery S.B., Astakhova T., Bilenky M., et al. Genome Research, 2004 | |
Assessment and integration of publicly available SAGE, cDNA microarray, and oligonucleotide microarray expression data for global coexpression analyses | Cross-platform analysis of gene expression from 1202 cDNA microarrays, 242 SAGE libraries, and 667 Affymetrix arrays. Compared co-expression prediction reliability and agreement across platforms using Gene Ontology validation. Affymetrix showed highest biological relevance. | Griffith O.L., Pleasance E.D., Fulton D.L., et al. Genomics, 2005 | |
An application of peer-to-peer technology to the discovery, use and assessment of bioinformatics programs | Development of an open-source peer-to-peer system for remote access and execution of bioinformatics tools. Enables distributed computing and sharing of algorithms through BioPerl and Java interfaces. Integrated with Ensembl data retrieval. | Montgomery S.B., Fu T., Guan J., Lin K., Jones S.J. Nature Methods, 2005 |